Transcription factor binding site
Transcription factor binding site是實驗能證明的區域,這段序列能與transcription factor 結合,進而開啟或關閉基因表現。本文沒有要強調Transcription factor binding site如何尋找或進行實驗驗證,僅介紹如何描述這段transcription factor binding site。
字串描述
最簡單的描述方法是CCATATATAG 這樣的單純字串描述,如下圖所示 (https://jaspar.genereg.net/sites/MA0001.1/)
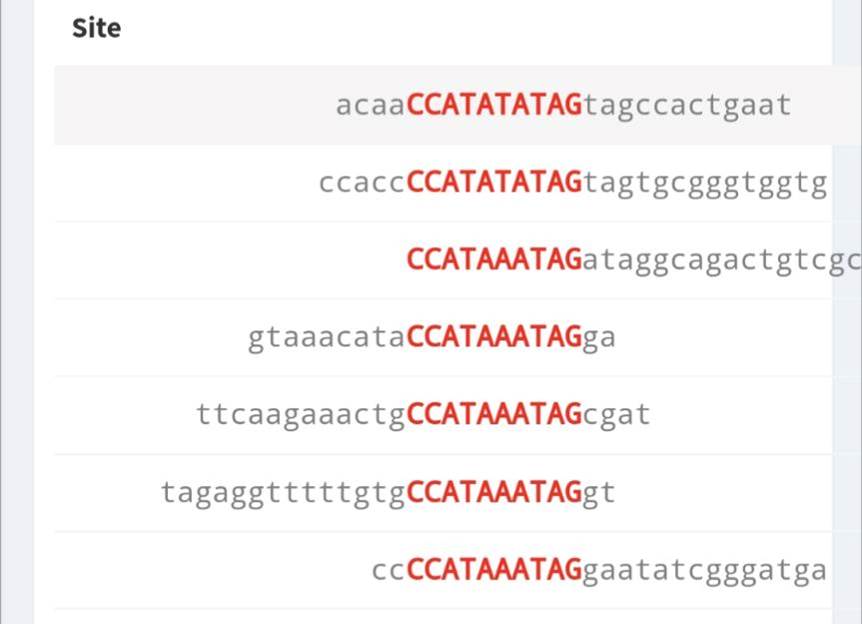
這種顯示方式最直觀,也最好利用,大家可以想像只要使用Ctrl + F或grep等方式進行搜尋。
正規表示法描述
有些位置如果有超過一種以上的DNA出現,這時候單純用ATCGU表現就不太方便了。目前IUPAC有訂立一系列標準用字去描述這些可能性,請參考 https://en.m.wikipedia.org/wiki/Nucleic_acid_notation ,就像R代表purine,包含AG。而Y代表Pyrimidine,包含CT等等。
Position weight matrix (PWM)
其實,transcription factor binding site的專一性很少有達到需每一個base都正確才有效能。通常transcription factor binding site是由一堆可能性堆疊而成,它描述了每個位置出現的ATCG頻率。以下圖左邊為例,直的column 代表每個位置,而橫的row代表ATCG,所以矩陣裡面的數值就是每個位置 (column)裡,ATCG出現的頻率。以第一個位置而言,看起來C出現的頻率最高,但Q或G仍有出現之可能。
| PMW | DNAlogo |
|---|---|
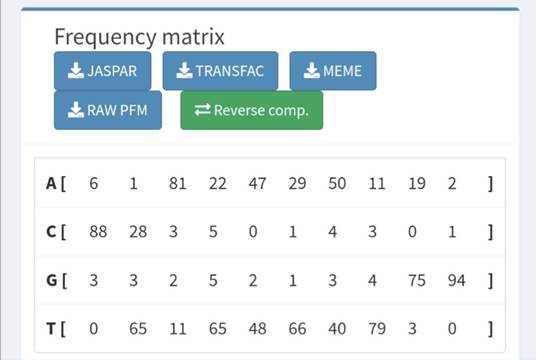 |  |
Ref. https://jaspar.genereg.net/matrix/MA0001.1/?revcomp=1
通常這種描述太數學了,如果想更視覺化一點,我們會把這些序列轉成DNAlogo的型態來描述,如上圖右側所示,因為第一個位置C出現的機率遠大約於其他核酸,所以就會看到一根很大的C。
好,那position weighted matrix看起來最實際,但該怎樣用它來搜尋全基因體,或是promoter region呢?
MEME package FIMO找序列
簡單暴力解,MEME package 裡面的FIMO專做這件事情 (https://meme-suite.org/meme/tools/fimo),只要有PWM,並改成MEME家的格式,餵給FIMO,再給一段序列,FIMO就能幫忙尋找這段PWM是否存在給定序列裡了。
這樣有簡單的輸出結果可以參考 https://meme-suite.org/meme/doc/examples/fimo_example_output_files/fimo.html 。MEME package 最貼心的是,他會在最下面列出現在使用的指令是什麼,你可以使用這指令在自己的電腦上重現這些結果
# Command line:
Fimo –oc fimo_example_output_files –parse-genomic-coord some_vertebrates.meme mm9_tss_500bp_sampled_1000.fna
# Settings:
Output_directory = fimo_example_output_files
MEME file name = some_vertebrates.meme sequence file name = mm9_tss_500bp_sampled_1000.fna
Background file name = --nrdb--
alphabet = DNA
max stored scores = 100000
Allow clobber = true
compute q-values = true parse genomic coord. = true
Text only = false
scan both strands = true max strand = false
Threshold type = p-value
output theshold = 0.0001
pseudocount = 0.1
Alpha = 1 verbosity = 2
MEME package MEME建立PWM
那如何建立PMW呢?最棒的方法是用multiple sequence alignment,真實的看到了某堆序列裡,真的有一段conserved sequence (高度保留序列),我們再拿這個高度保留序列製成PMW。但這挑戰性極高,DNA multiple sequence alignment 沒那麼好做,所以簡單做法是透過生物實驗,像是RNA sequence找出coexpression network,或是用kmean把具有一致expression patterns 的基因們打撈出來,甚至做chip seq,直接測量蛋白質與DNA的結合。最後再拿這些資料給MEME,讓他幫忙找在這些序列裡是不是真的藏有一些短片段。
注意:MEME定義的PMW motif有兩大前提假設,第一是gapless,也就是禁止出現gap。第二,長度要一致。也就是說MEME如果發現兩個長度不一致的motif,它會直接定義成兩個motif
實際範例:https://meme-suite.org/meme/doc/examples/meme_example_output_files/meme.html?man_type=web
meme lex0.fna -oc meme_example_output_files -dna -mod zoops -nmotifs 3 -revcomp -mpi
背後原理
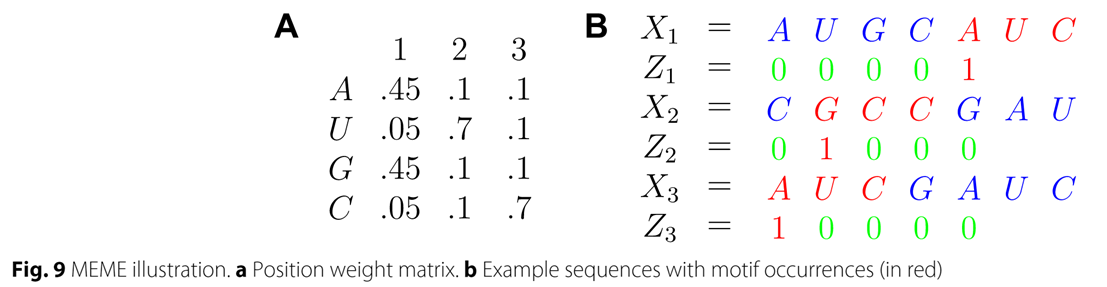
MEME背後是進行EM (Expectation-Maximization Algorithm)。實際範例可以參考 RNA motif discovery: a computational overview | Biology Direct | Full Text (biomedcentral.com)。下圖A是一個典型的PMW,而圖B是實際應用。X代表序列,而Z代表有沒有搜尋到motif,0代表沒有,1代表有,且紅色的序列就代表了motif。由於第一個位置與許A與G的機率很高,所以可以看到A、G都能當成第一個序列